之前我们分享了一些机器学习在有机合成应用上的优秀案例“如虎添翼—AI为有机合成插上腾飞的翅膀”,内容来自德国哥廷根大学的Lutz Ackermann和浙江大学的XinHong 在Trends in Chemistry上发表的一篇综述 “When machine learning meets molecular synthesis”[1]。为建立质量高的模型,良好的数据库必不可少,理想的有机合成数据库需具有大规模、公开性、标准化等特点,能节省数据收集和整理的时间,本篇我们会介绍一些具代表性的有机合成机器学习数据库。
有机合成领域的文献数量不少,然而这些数据往往以冷数据形式收录,缺乏标准化整理,导致在建立模型时需要花大量的时间去收集和整理数据。因此,理想的有机合成数据库具有大规模、公开性、标准化等特点。根据不同的数据来源,数据库被分类成基于实验的数据库、基于计算的数据库和开源反应数据库。德国慕尼黑大学Mayr教授课题组通过Mayr方程实现了对亲核性与亲电性反应的定量标度并建立了数据库[2]。Mayr方程(图1A)由三个基于对速率常数的实验测量所得的经验参数组成,E为亲电参数,N为溶剂相关的亲核性参数, sN为溶剂相关的亲核敏感性参数。迄今为止,Mayr 的反应性参数数据库测量了 345 个亲电试剂和1250 个亲核试剂的参数。当中记录了132个碳正离子 、114个缺电子烯烃、9个硫 、5个氟 、4个氮 和 3个氯亲电体及78个其他碳亲电试剂 。亲核试剂则包括 530 个碳, 311 个氮、191 个氢、138个 氧、33 个硫和硒、29 个卤化物阴离子和 18 个磷亲核试剂。测得的反应性参数的分布接近正态分布(图1C),并基于置信度对数据库进行了分类。我们基于这个数据库可以预测超过 4,300,000 个反应组合的二级速率常数。
图1. Mayr's Database
NextMove通过文本挖掘技术,对美国专利中的有机合成反应作进一步标注,建立了USPTO 数据库[3],提高了数据的可用性。来自诺华生物医学研究所的Schneider课题组通过USPTO 数据库的标签进行挑选,整理出子集USPTO-50k数据库[4],图2是USPTO-50k涵盖的有机反应类型。
图2. USPTO-50k Database
随着理论计算的进步,科学家可通过计算化学研究有机化学,预测分子性质和反应性能。计算化学数据库通常比实验数据库的规模更大,数据更完整。來自巴塞尔大学的Lilienfeld课题组在B3LYP/6-31G(2df,p)的方法基组下计算了133 885 个化合物(最多含9 个重原子)的量化性质并建立了QM9数据库[5],QM9的数据分布和计算特征如图3所示。QM9已被广泛应用于机器学习中,包含化合物特性预测、合成性能预测等。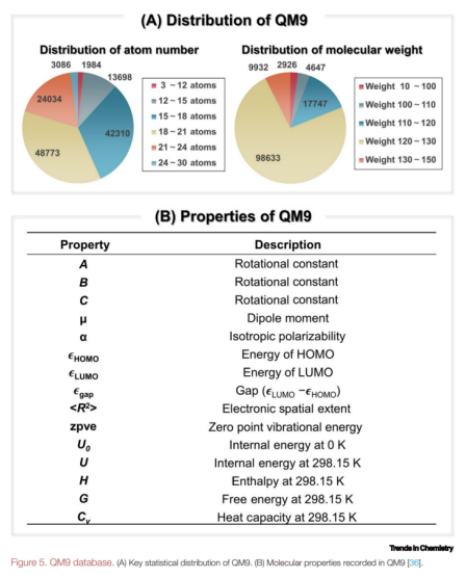
图3. QM9 Database
基于计算化学的数据库—杂环化合物C-H键官能化反应库
浙江大学的XinHong课题组基于DFT计算了环化合物C-H键官能化反应的能量势垒,建立了反应数据库(图4)[6],数据库涉及了201个芳烃取代模式和 13 个自由基,6114 个竞争位的势垒和 9438 个势垒差异。该数据库被用作训练了一个随机森林模型并展现出令人满意的选择性预测效能,表明了DFT 计算可以作为一种有用的数据增强策略。
图4. 杂环化合物C-H键官能化反应库
来自Relay Therapeutics的Steven M. Kearnes和麻省理工学院MIT的Connor W. Coley等人建立了开源反应数据库(Open Reaction Database, ORD)[7],一种用于建立和分享有机反应数据的公开访问架构和基础的设施,期望符合科学数据管理的FAIR原则,方便下游数据取用。ORD架构可支持传统实验反应,也可支持新兴的自动化高通量实验和流动化学等技术。图5为ORD当前可用的示例数据集,在GitHub上可获取(https://github.com/open-reaction-database)。
图5. ORD示例数据集
有机合成反应数据库涉及的层面广泛,由于研究领域发展已久且数据量大,实验记录已从传统的纸本发展到今天的电子实验记录本,但仍未规范标准化可用于机器学习的数据记录格式,为数据库的收集和整理带来困难。本文介绍的数据库尝试整理了实验、计算化学等数据,考虑到数据未来可用作机器学习摸型的特征制作了标签。近年更出现ORD架构,期望能广泛用于日后的实验数据记录中,可见有机合成数据库建立已有雏形,为虚拟信息发展迅速的未来作好准备。
[1] Oliveira J C A, Frey J, Zhang S Q, et al. (2022). When machine learning meets molecular synthesis. Trends in Chemistry.[2] Mayr, H. et al. (2003) π-Nucleophilicity in carbon–carbon bondforming reactions. Acc. Chem. Res. 36, 66–77[3] Schneider, N. et al. (2016) Big data from pharmaceutical patents: a computational analysis of medicinal chemists’ bread and butter. J. Med. Chem. 59, 4385–4402[4] Schneider, N. et al. (2016) What’s what: the (nearly) definitive guide to reaction role assignment. J. Chem. Inf. Model. 56, 2336–2346[5] Ramakrishnan, R. et al. (2014) Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 1, 140022[6] Li, X. et al. (2020) Predicting regioselectivity in radical C–H functionalization of heterocycles through machine learning. Angew. Chem. Int. Ed. 59, 13253–13259[7] Kearnes, S.M. et al. (2021) The Open Reaction Database. J. Am. Chem. Soc. 143, 18820–1882
感兴趣的读者,可以添加小邦微信(zhiyaobang2020)加入读者实名讨论微信群。添加时请主动注明姓名-企业-职位/岗位 或
姓名-学校-职务/研究方向。











