绝大多数深度学习是在欧几里得数据上进行的,几何深度学习旨在构建可以从非欧几里得数据[几何对象如网格、空间点云、曲面等]中,整合和处理对称信息的神经网络。
在三维分子表示的背景下,对称性指的是旋转、平移、映射,以及相应的分子性质在这种转换下的行为,将对称信息(也称为几何先验)引入神经网络架构,实现了对3D分子图的有效学习,并已成为分子几何深度学习的主要方向之一。
基于几何深度学习的结构药物设计在药物发现过程中得到了很好的应用。

基于几何深度学习的大分子结构的3D表示可以用于(i)分子属性预测(如结合亲和力、蛋白质功能和pose打分),(ii) 结合位点和界面预测如小分子结合位点和蛋白质-蛋白质界面,(iii)结合pose生成和分子对接(如配体-蛋白质和蛋白质-蛋白质结合),以及(iv)基于结构的小分子配体的从头设计。
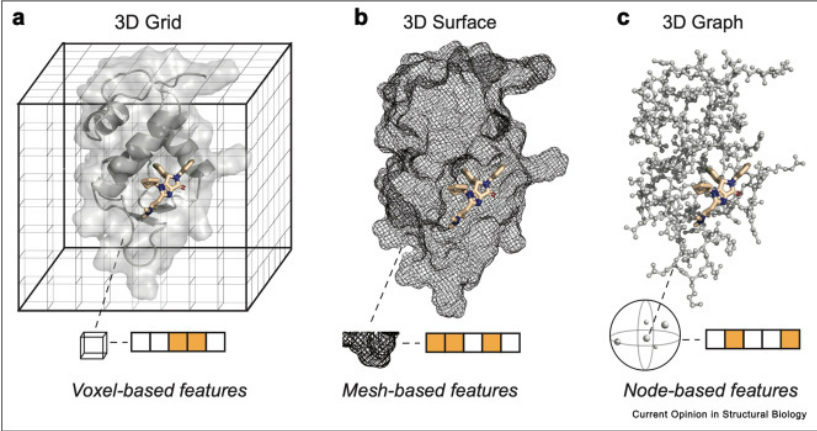
三种最普遍的大分子几何数据表征:3D网格、3D曲面和3D图。这三种表征法具有独特的几何形状和对称性。
3D Grid由欧几里得数据结构定义,该数据结构由3D空间中的Voxels组成。这种欧几里得几何以网格的各个Voxel为特征,具有固定的邻域几何。
3D Surface由描述网格坐标(网格空间)3D排列的多边形(面)组成。多边形可以根据它们的化学特征和由网格的局部几何形状定义的几何特征来区分。
3D Graph由非欧几里得数据结构定义,该结构由节点(由单个原子表示)及其边组成。这些边由相邻节点定义。
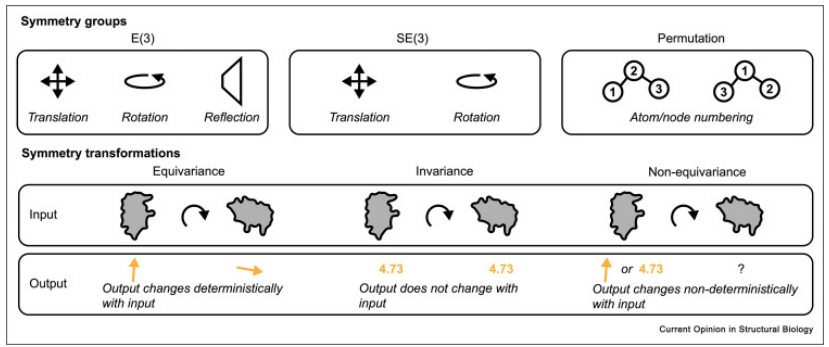
基于结构的药物设计相关的对称群(Symmetry groups)和变换(Symmetry transformations)用于结构药物设计。对称变换(下图),可以显示为下面灰色的部分即蛋白质结构的旋转,作为几何深度学习的输入。输出结构可以预测为向量(用黄色箭头显示)也可以是标量(用黄色数字显示)。
基于结构的药物设计的几何深度学习方法概述,方法根据任务(行)和大分子表示(列)放置。
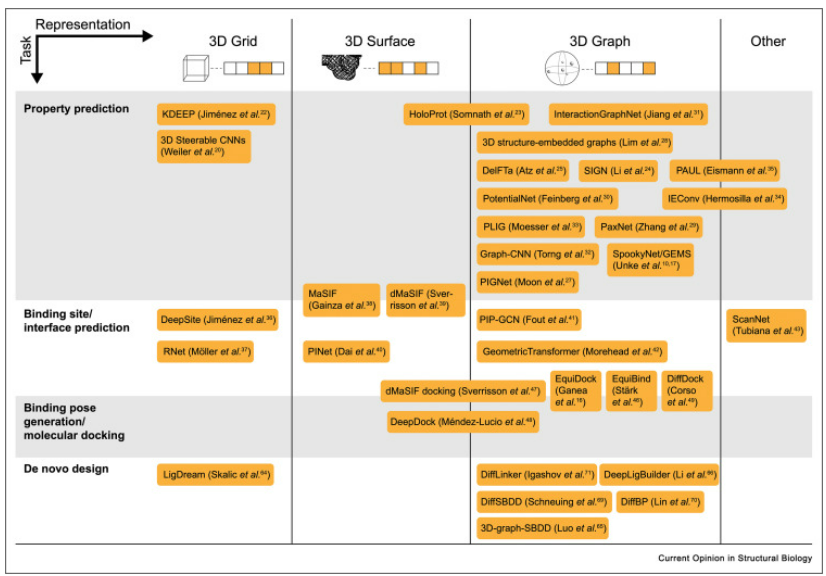
本文关注基于3D Surface的方法在药物发现过程中的应用。
在结合亲和力和蛋白质功能预测的方面,HoloProt通过组合基于序列、surface和结构的图的表示来编码不同长度尺度的蛋白质。Surface-level graph使用三角化蛋白质表面上的节点,并用物理化学和几何特征进行注释。
在结合位点/界面预测方法方面,MaSIF及其可微分方法dMaSIF用于大分子表面表征。基于surface的方法描述了蛋白质上各个点的测地线空间中,点之间的距离对应于它们之间沿表面的路径长度,而不是欧几里得距离。在三步过程中,surface被分解为单独的小块。每个patch中的点都具有几何和化学属性。测地线卷积将这些特征转换为用于下游任务的数值向量,前两个步骤需要在原始实现中进行昂贵的计算。
而dMaSIF是端到端可微的,直接在原子类型和坐标上运行。PINet使用受物理启发的几何深度学习网络,通过学习surface形状和物理化学特性的互补性来识别两种相互作用的蛋白质之间的界面区域。由于此网络架构中缺乏旋转不变性,因此需要使用输入结构的随机旋转来增强数据。
在对接pose预测方面,EquiDock使用SE(3)等变消息传递网络与optimal transport相结合去预测蛋白蛋白刚性结合pose,EquiBind将该方法扩展到柔性小分子配体和蛋白质结构对接。
dMaSIF,将SE(3)等变图神经网络与原子点云的表面指纹相结合,以估计两个结合蛋白质的表面形状互补性,用于刚性对接预测。
DeepDock用几何深度学习方法,通过将结合位点表面表示为多边形网格并将配体表示为分子图,预测配体和蛋白质之间成对节点距离上的概率分布来预测小分子结合pose。MaSIF是源于下面2019年发表在Nature子刊的文章,文章第一次成功将几何深度学习用于基于3D surface的分子相互作用指纹学习。

分子表面数据是在测地线空间中描述的,这意味着两点之间的距离对应于沿着表面的点之间的”walking“距离。在高度不规则的蛋白质表面(例如,具有深口袋)中,测地距离可能比欧几里得距离大的多。
首先,MaSIF将曲面分解为具有固定测地半径的重叠径向patch[在网格的每个vertex,提取测地半径r为9埃或r=12埃的patch],在patch内的每个点(vertex)用几何特征[曲面形状指数, 依赖于距离的曲率]和化学特征[疏水指数,连续静电,自由电子和质子供体的位置]的数组表示。输入特征(化学和几何)不是学习的,是从分子表面预先计算的.
图a. 左边是刻有相互作用指纹的蛋白质表面的概念表示,surface 特征可能揭示了其潜在的生物分子相互作用。右,将曲面分割为MaSIF中使用的测地线半径的重叠径向patches. 图b. patches包括映射在蛋白质表面上的几何和化学特征。然后,MaSIF学习将曲面块的输入特征嵌入到数字向量的描述符中(图d)。每个描述符都使用依赖于应用的神经网络层进一步处理。网络是端到端训练的,这意味着中间patch描述符不是通用的,而是针对特定任务进行优化的。
图c. 测地线极坐标,用于映射patch内的特征的位置。径向坐标(radial coordinate)表示到patch的中心的测地距离,角坐标(angular coordinate)相对于patch中心的随机方向计算。表面的几何结构(如,表面内口袋的深度)通过几何特征和测地极坐标隐含的表示。图d. MaSIF使用几何深度学习工具将特定的神经网络应用于数据。使用包含可重复使用的构建块(测地卷积层)的神经网络架构为每个patch计算指纹描述符.
MaSIF可以应用在(1)配体口袋相似性比较(MaSIF-ligand);(2) 蛋白-蛋白相互作用位点的预测(MaSIF-site);(3) surfaces快速扫描,利用表面指纹来预测蛋白质-蛋白质复合物结构(MaSIF-search)。
图e. MaSIF可适用于多个预测任务。
Youtube视频:
同一批作者将该表征方法用于蛋白质相互作用的从头设计,已在2023年4月26日发表在了Nature顶刊。

作者设计了三阶段的计算方法:(1)使用MaSIF-site预测具有高结合倾向的目标包埋的界面位点,图a;(2)基于表面指纹的搜索互补结构基序(结合种子)显示结合目标位点所需的特征,称之为MaSIF-seed的协议,图a,b;(3)使用已建立的转移[transplantation]技术将binding种子转移到蛋白质支架上,以在设计的结合界面上找到稳定的和额外的contact。
从头设计蛋白质与学习表面指纹的相互作用。图a. 指纹生成示意图。蛋白质结合位点在空间上嵌入为向量指纹[Vector fingerprints],蛋白质表面被分解为重叠的径向patch,并且在天然相互作用的蛋白质对上训练几何深度学习用以学习嵌入指纹,以便将互补指纹放置在相似的空间区域中。作者展示了缩小到三维空间中的指纹,绿色框突出显示了互补指纹区域。图b. MaSIF-seed一种识别新结合种子的方法。MaSIF-site根据形成包埋界面的倾向识别目标patch。使用MaSIF-seed,在目标patch和大型数据库(约4.02亿patchs)中的所有指纹之间评估指纹互补性;随后对指纹进行排序,对top patch进行对齐和重新评分,以便对种子候选patch进行更精确的评估。图c. scaffold搜索、种子转移和界面重新设计。选定的种子被转移到蛋白质scaffold上,界面的其余部分使用Rosetta重新设计。top设计被选择并通过实验进行测试。作者计算设计了几种从头设计蛋白质结合剂来结合四种蛋白质靶标:SARS-CoV-2 刺突蛋白、PD-1、PD-L1 和 CTLA-4。一些设计经过实验优化,而其他设计则完全在计算机中生成,达到纳摩尔亲和力,结构和突变表征显示出高度准确的预测。
总的来说,以surface为中心的方法捕获了分子识别的物理和化学决定因素,从而实现了蛋白质相互作用的从头设计方法,更广泛地说,具有功能的人造蛋白质。下面的nature子刊将基于surface的表面特征和与之适用的几何深度学习方法也被用于靶标和活性分子预测的场景中。

2023年4月18日发表的Nature子刊文章将无参数的几何深度学习高度可信地预测和区分设计核酸、脂质、离子和小分子的界面。为发现未探索的生物学提供了新的机会。

下面2022年5月30日Nauture子刊用一种端到端、可解释的几何深度学习模型,可直接从 3D 结构中学习特征。并在蛋白质-蛋白质和蛋白质-抗体结合位点上验证了其准确性。

几何深度学习同样也用在三维小分子的空间表征中。

参考文献
[1] Clemens Isert, Kenneth Atz, Gisbert Schneider,Structure-based drug design with geometric deep learning,Current Opinion in Structural Biology,Volume 79,2023,102548,ISSN 0959-440X,https://doi.org/10.1016/j.sbi.2023.102548.
[2] Gainza, P., Sverrisson, F., Monti, F. et al. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat Methods 17, 184–192 (2020). https://doi.org/10.1038/s41592-019-0666-6.
[3] Gainza, P., Wehrle, S., Van Hall-Beauvais, A. et al. De novo design of protein interactions with learned surface fingerprints. Nature 617, 176–184 (2023). https://doi.org/10.1038/s41586-023-05993-x.
[4] Krapp, L.F., Abriata, L.A., Cortés Rodriguez, F. et al. PeSTo: parameter-free geometric deep learning for accurate prediction of protein binding interfaces. Nat Commun 14, 2175 (2023). https://doi.org/10.1038/s41467-023-37701-8.
[5] Tubiana, J., Schneidman-Duhovny, D. & Wolfson, H.J. ScanNet: an interpretable geometric deep learning model for structure-based protein binding site prediction. Nat Methods 19, 730–739 (2022). https://doi.org/10.1038/s41592-022-01490-7.
[6] Méndez-Lucio, O., Ahmad, M., del Rio-Chanona, E.A. et al. A geometric deep learning approach to predict binding conformations of bioactive molecules. Nat Mach Intell 3, 1033–1039 (2021). https://doi.org/10.1038/s42256-021-00409-9.
[7] Atz, K., Grisoni, F. & Schneider, G. Geometric deep learning on molecular representations. Nat Mach Intell 3, 1023–1032 (2021). https://doi.org/10.1038/s42256-021-00418-8.
感兴趣的读者,可以添加小邦微信(zhiyaobang2020)加入读者实名讨论微信群。添加时请主动注明姓名-企业-职位/岗位 或 姓名-学校-职务/研究方向。










